The Covid Delta variant adapts itself to humans and hides what it has done, preparing itself to propagate and infect others.
It's power and secrecy combine to make it especially dangerous because it is so difficult to understand. JL
Megan Scudellari reports in Nature:
Scientists have discovered key adaptations that help the virus to grab
on to human cells with surprising strength and then hide itself once
inside. Later, as it leaves cells, SARS-CoV-2 executes a crucial
processing step to prepare its particles for infecting even more human
cells. These are some of the tools that have enabled the virus to spread
so quickly and claim millions of lives. “That’s why it’s so difficult
to control.”
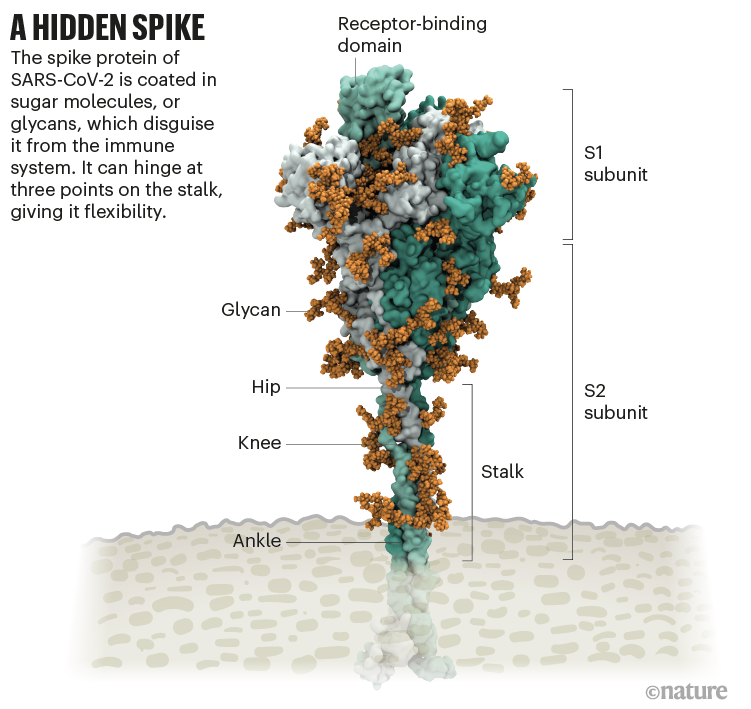
The coronavirus sports a luxurious sugar coat. “It’s striking,” thought Rommie Amaro, staring at her computer simulation of one of the trademark spike proteins of SARS-CoV-2, which stick out from the virus’s surface. It was swathed in sugar molecules, known as glycans.
“When you see it with all the glycans, it’s almost unrecognizable,” says Amaro, a computational biophysical chemist at the University of California, San Diego.
Many viruses have glycans covering their outer proteins, camouflaging them from the human immune system like a wolf in sheep’s clothing. But last year, Amaro’s laboratory group and collaborators created the most detailed visualization yet of this coat, based on structural and genetic data and rendered atom-by-atom by a supercomputer. On 22 March 2020,she posted the simulation to Twitter. Within an hour, one researcher asked in a comment: what was the naked, uncoated loop sticking out of the top of the protein?
Amaro had no idea. But ten minutes later, structural biologist Jason McLellan at the University of Texas at Austin chimed in: the uncoated loop was a receptor binding domain (RBD), one of three sections of the spike that bind to receptors on human cells (see ‘A hidden spike’).
Source: Structural image from Lorenzo Casalino, Univ. California, San Diego (Ref. 1); Graphic: Nik Spencer/Nature
In Amaro’s simulation, when the RBD lifted up above the glycan cloud,two glycans swooped in to lock it into place, like a kickstand on a bicycle. When Amaro mutated the glycans in the computer model, the RBD collapsed. McLellan’s team built a way to try the same experiment in the lab, and by June 2020, the collaborators had reported that mutating the two glycans reduced the ability of the spike protein to bind to a human cell receptor1— a role that no one has previously recognized in coronaviruses, McLellan says. It’s possible that snipping out those two sugars could reduce the virus’s infectivity, says Amaro, although researchers don’t yet have a way to do this.
Since the start of the COVID-19 pandemic, scientists have been developing a detailed understanding of how SARS-CoV-2 infects cells. By picking apart the infection process, they hope to find better ways to interrupt it through improved treatments and vaccines, and learn why the latest strains, such as the Delta variant,are more transmissible.
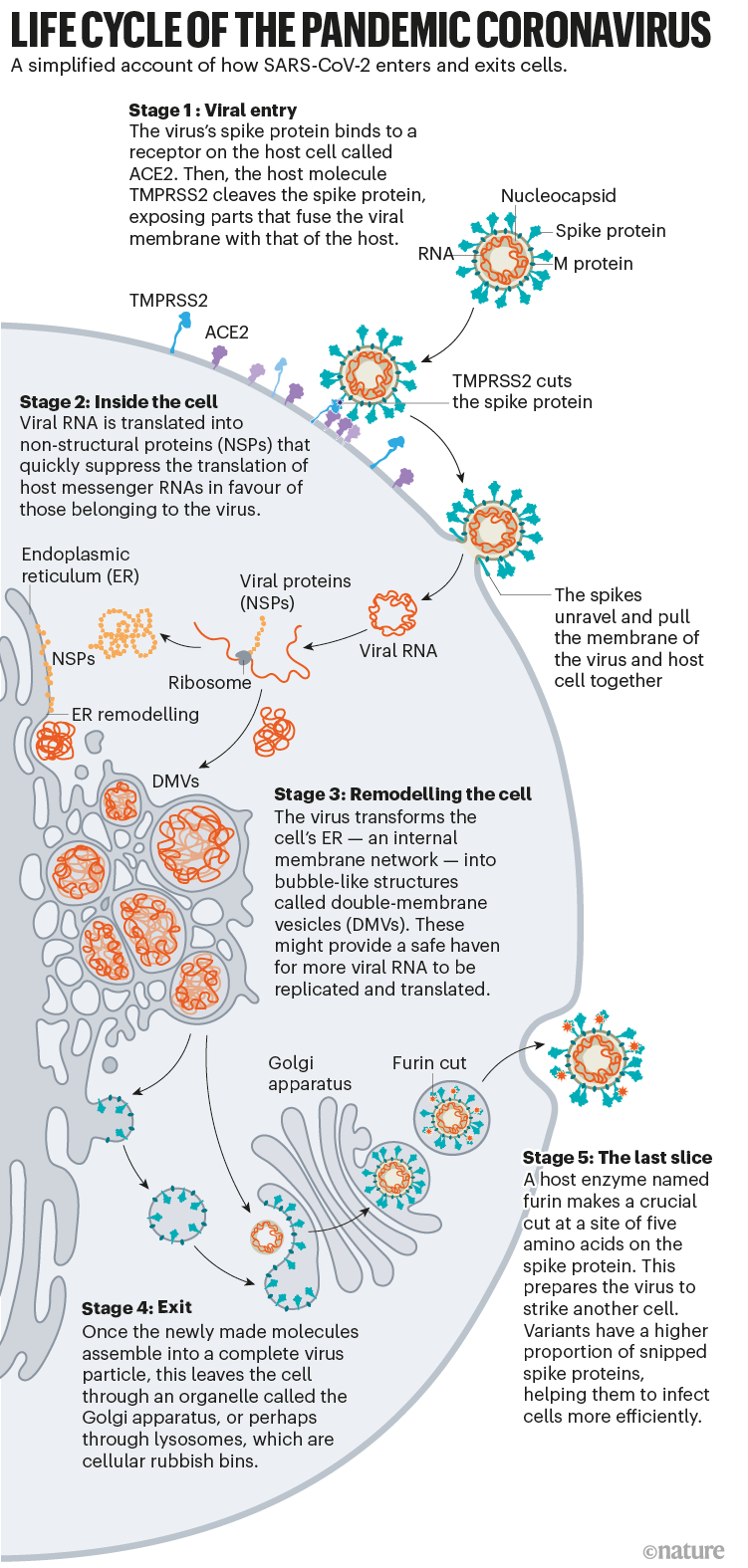
What has emerged from 19 months of work, backed by decades of coronavirus research, is a blow-by-blow account of how SARS-CoV-2 invades human cells (see ‘Life cycle of the pandemic coronavirus’). Scientists have discovered key adaptations that help the virus to grab on to human cells with surprising strength and then hide itself once inside. Later, as it leaves cells, SARS-CoV-2 executes a crucial processing step to prepare its particles for infecting even more human cells. These are some of the tools that have enabled the virus to spread so quickly and claim millions of lives. “That’s why it’s so difficult to control,” says Wendy Barclay, a virologist at Imperial College London.
It starts with the spikes. Each SARS-CoV-2 virion (virus particle) has an outer surface peppered with 24–40 haphazardly arranged spike proteins that are its key to fusing with human cells2. For other types of virus, such as influenza, external fusion proteins are relatively rigid. SARS-CoV-2 spikes, however, are wildly flexible and hinge at three points, according to work published in August 2020 by biochemist Martin Beck at the Max Planck Institute of Biophysics in Frankfurt, Germany, and his colleagues3.
That allows the spikes to flop around, sway and rotate, which could make it easier for them to scan the cell surface and for multiple spikes to bind to a human cell. There are no similar experimental data for other coronaviruses, but because spike-protein sequences are highly evolutionarily conserved, it is fair to assume the trait is shared, says Beck.
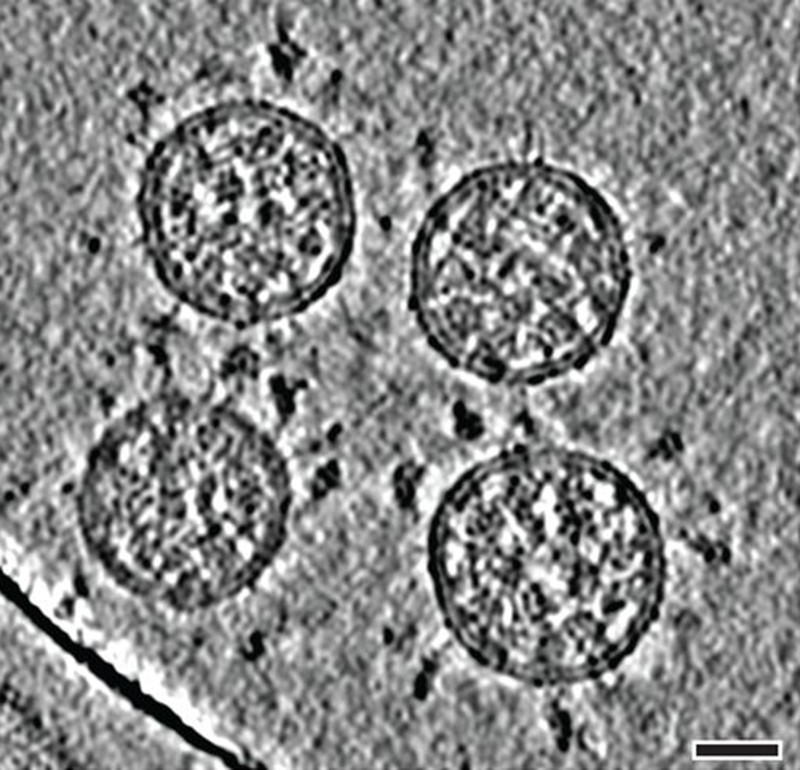
Cryo-electron tomography images of SARS-CoV-2 virions. (Scale bar: 30 nanometres.)Credit: B. Turoňováet al./Science
Early in the pandemic, researchers confirmed that the RBDs of SARS-CoV-2 spike proteins attach to a familiar protein called the ACE2 receptor, which adorns the outside of most human throat and lung cells. This receptor is also the docking point for SARS-CoV, the virus that causes severe acute respiratory syndrome (SARS). But compared with SARS-CoV, SARS-CoV-2 binds to ACE2 an estimated 2–4 times more strongly4, because several changes in the RBD stabilize its virus-binding hotspots5.
Worrying variants of SARS-CoV-2 tend to have mutations in the S1 subunit of the spike protein, which hosts the RBDs and is responsible for binding to the ACE2 receptor. (A second spike subunit, S2, prompts viral fusion with the host cell’s membrane.)
The Alpha variant, for example, includes ten changes in the spike-protein sequence, which result in RBDs being more likely to stay in the ‘up’ position6. “It is helping the virus along by making it easier to enter into cells,” says Priyamvada Acharya, a structural biologist at the Duke Human Vaccine Institute in Durham, North Carolina, who is studying the spike mutations.
The Delta variant, which is now spreading around the world, hosts multiple mutations in the S1 subunit, including three in the RBD that seem to improve the RBD’s ability to bind to ACE2 and evade the immune system7.
Restricted entry
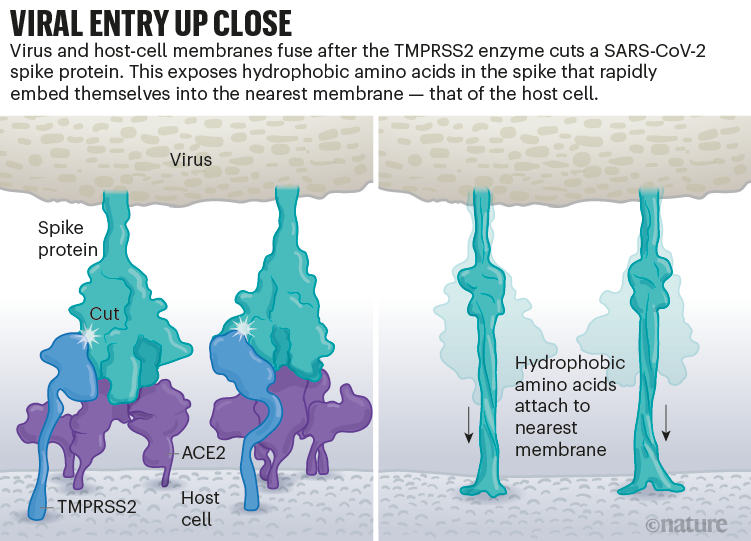
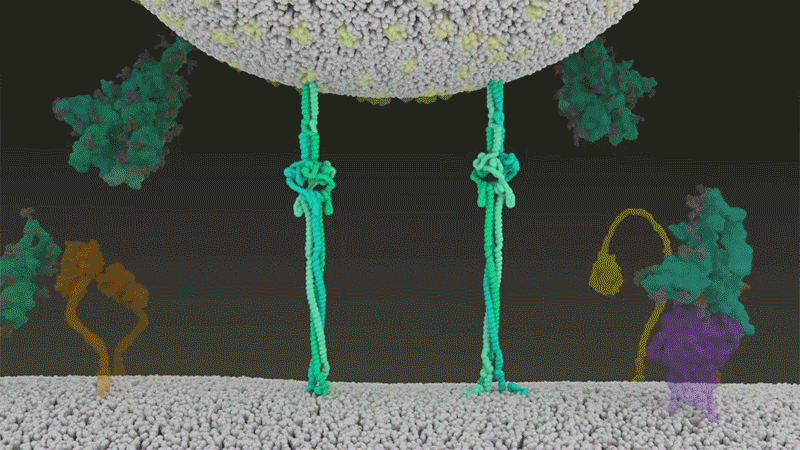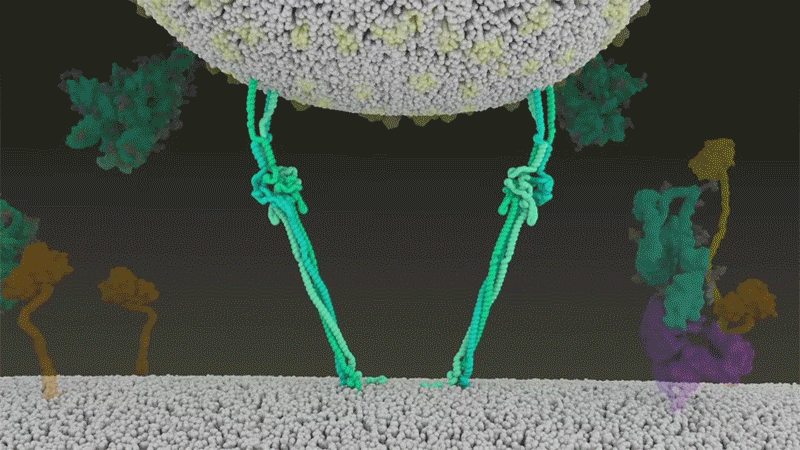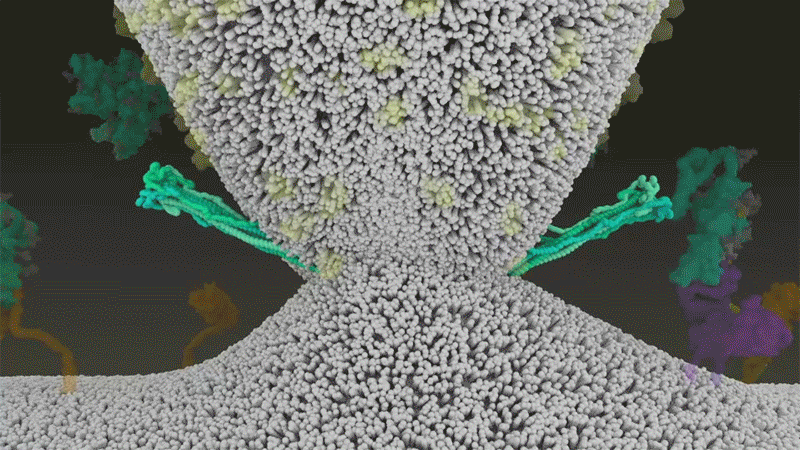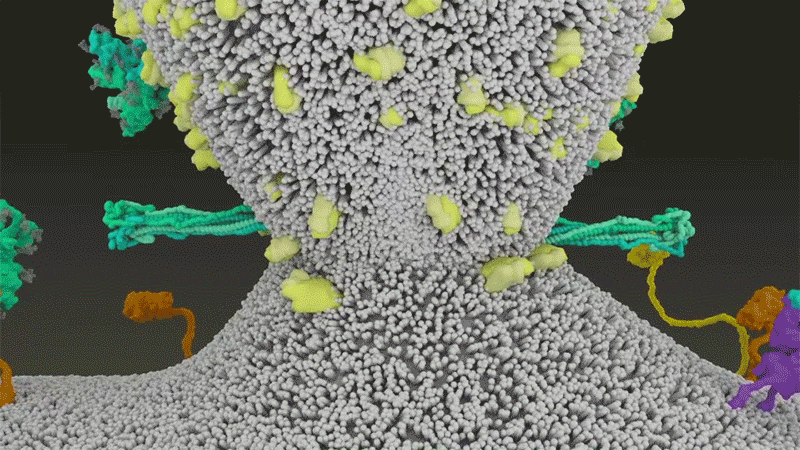
Once the viral spikes bind to ACE2, other proteins on the host cell’s surface initiate a process that leads to the merging of viral and cell membranes (see ‘Viral entry up close’).
The virus that causes SARS, SARS-CoV, uses either of two host protease enzymes to break in: TMPRSS2 (pronounced ‘tempress two’) or cathepsin L. TMPRSS2 is the faster route in, but SARS-CoV often enters instead through an endosome — a lipid-surrounded bubble — which relies on cathepsin L. When virions enter cells by this route, however, antiviral proteins can trap them.
SARS-CoV-2 differs from SARS-CoV because it efficiently uses TMPRSS2, an enzyme found in high amounts on the outside of respiratory cells. First, TMPRSS2 cuts a site on the spike’s S2 subunit8. That cut exposes a run of hydrophobic amino acids that rapidly buries itself in the closest membrane — that of the host cell. Next, the extended spike folds back onto itself, like a zipper, forcing the viral and cell membranes to fuse.
The virus then ejects its genome directly into the cell. By invading in this spring-loaded manner, SARS-CoV-2 infects faster than SARS-CoV and avoids being trapped in endosomes, according to work published in April by Barclay and her colleagues at Imperial College London9.
The virus’s speedy entry using TMPRSS2 explains why the malaria drug chloroquine didn’t work in clinical trials as a COVID-19 treatment, despite early promising studies in the lab10. Those turned out to have used cells that rely exclusively on cathepsins for endosomal entry. “When the virus transmits and replicates in the human airway, it doesn’t use endosomes, so chloroquine, which is an endosomal disrupting drug, is not effective in real life,” says Barclay.
The discovery also points to protease inhibitors as a promising therapeutic option to prevent a virus from using TMPRSS2, cathepsin L or other proteases to enter host cells. One TMPRSS2 inhibitor, camostat mesylate, which is approved in Japan to treat pancreatitis, blocked viral entry into lung cells8, but the drug did not improve patients’ outcomes in an initial clinical trial11.
“From my perspective, we should have such protease inhibitors as broad antivirals available to fight new disease outbreaks and prevent future pandemics at the very beginning,” says Stefan Pöhlmann, director of the Infection Biology Unit at the German Primate Center in Göttingen, who has led research on ACE2 binding and the TMPRSS2 pathway.
Deadly competition
The next steps of infection are murkier. “There are a lot more black boxes once you are inside the cell,” says chemist Janet Iwasa at the University of Utah in Salt Lake City, who is developing an annotated animation of the viral life cycle. “There’s more uncertainty, and competing hypotheses.”
After the virus shoots its RNA genome into the cell, ribosomes in the cytoplasm translate two sections of viral RNA into long strings of amino acids, which are then snipped into 16 proteins, including many involved in RNA synthesis. Later, more RNAs are generated that code for a total of 26 known viral proteins, including structural ones used to make new virus particles, such as the spike, and other accessory proteins. In this way, the virus begins churning out copies of its own messenger RNA. But it needs the cell’s machinery to translate those mRNAs into proteins.
Coronaviruses take over that machinery in many ways. Virologist Noam Stern-Ginossar and her team at the Weizmann Institute of Science in Rehovot, Israel, zoomed in on three mechanisms by which SARS-CoV-2 suppresses the translation of host mRNA in favour of its own. None are exclusive to this virus, but the combination, speed and magnitude of the effects seem unique, says Stern-Ginossar.
First, the virus eliminates the competition: viral protein Nsp1, one of the first proteins translated when the virus arrives, recruits host proteins to systematically chop up all cellular mRNAs that don’t have a viral tag. When Stern-Ginossar’s team put that same tag on the end of a host mRNA, the mRNA was not chopped up12.
Second, infection reduces overall protein translation in the cell by 70%. Nsp1 is again the main culprit, this time physically blocking the entry channel of ribosomes so mRNA can’t get inside, according to work from two research teams13,14. The little translation capacity that remains is dedicated to viral RNAs, says Stern-Ginossar.
Finally, the virus shuts down the cell’s alarm system. This happens in numerous ways, but Stern-Ginossar’s team identified one clear mechanism for SARS-CoV-2: the virus prevents cellular mRNA from getting out of the nucleus, including instructions for proteins meant to alert the immune system to infection. A second team confirmed this finding, and again pointed to Nsp1: the protein seems to jam up exit channels in the nucleus so nothing can escape15.
Because gene transcripts can’t get out of the nucleus, the infected cells don’t release many interferons — these are signalling proteins that alert the immune system to the presence of a virus. SARS-Cov-2 is particularly efficient at shutting down this alarm system: compared with other respiratory viruses, including SARS-CoV and respiratory syncytial virus, SARS-CoV-2 infection induces significantly lower levels of interferons16. And this June, researchersreported mutations in the Alpha variantthat seem to enable it to subdue interferon production even more efficiently17.
“It’s clear that SARS-CoV-2 is a very fast virus that has a unique ability to prevent our immune system from recognizing and combating infection in the first stages,” says Stern-Ginossar. By the time the immune system does realize there is a virus, there is so much of it that immune-response proteins sometimes flood the bloodstream at a faster rate than normal — which can cause damage. Doctors saw early in the pandemic that some people with COVID-19 who become very ill are harmed by an overactive immune response to SARS-CoV-2, as well as by the virus itself. Some proven treatmentswork by dampening down this immune response.
Renovation station
Once the virus has taken over host translation, it starts a home makeover, extensively remodelling the interior and exterior of the cell to its needs.
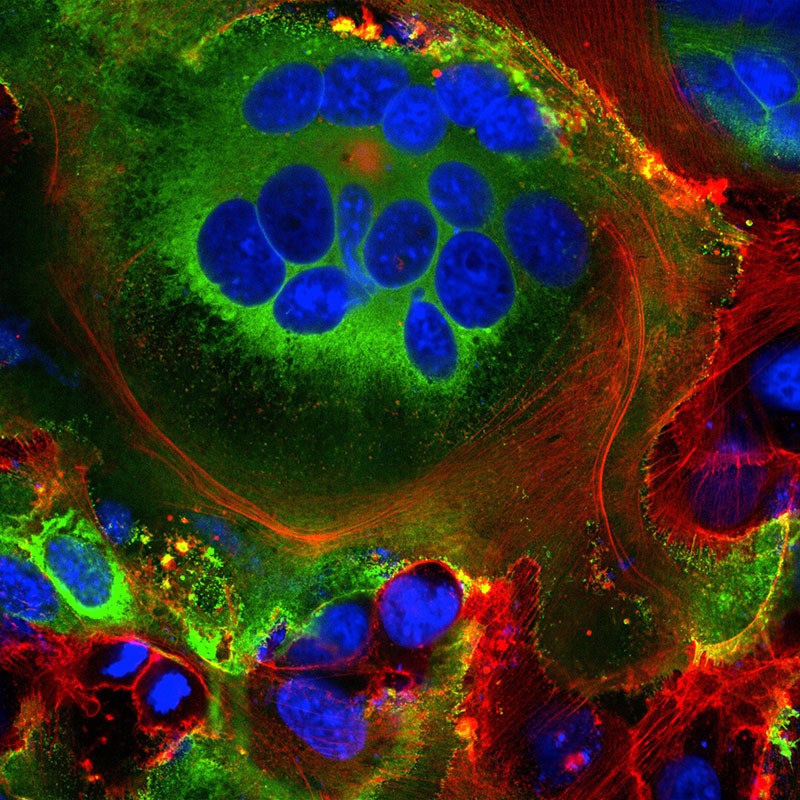
First, some of the newly made viral spike proteins travel to the surface of the cell and poke out of the host-cell membrane. There, they activate a host calcium-ion channel, which expels a fatty coating onto the outside of the cell — the same coating found on cells that naturally fuse together, such as muscle cells. At this point, the infected cell fuses to neighbouring cells expressing ACE2, developing into massive individual respiratory cells filled with up to 20 nuclei.
Fused cell structures (syncytia) seen in cells expressing the SARS-CoV-2 spike protein (green). Nuclei are in blue and the cell skeleton is in red.Credit: Mauro Giacca
These fused structures, called syncytia, are induced by viral infections such as HIV and herpes simplex virus, but not by the SARS virus, says molecular biologist Mauro Giacca at King’s College London, who led the team that published the finding in April18. He hypothesizes that forming syncytia allows infected cells to thrive for long periods of time, churning out more and more virions. “This is not a hit-and-run virus,” he says. “It persists.” A second team, led by researcher Qiang Sun at the Chinese Academy of Medical Sciences in Beijing, found that some COVID-19-infected cells even form syncytia with lymphocytes — one of the body’s own immune cells19. This is a known mechanism of immune evasion by tumour cells, but not by viruses. It suggests that infected cells avoid immune detection by simply grabbing on to and merging with nearby immune scouts.
On the inside of the cell, even more change is occurring. Like other coronaviruses, SARS-CoV-2 transforms the long, thin endoplasmic reticulum (ER), a network of flat membranes involved in protein synthesis and transport, into double-membrane spheres, as if the ER were blowing bubbles. These double-membrane vesicles (DMVs) might provide a safe place for viral RNA to be replicated and translated, shielding it from innate immune sensors in the cell, but that hypothesis is still being investigated.
Proteins involved in making DMVs could be good drug targets, because they seem to be necessary for viral replication. For instance, a host protein, TMEM41B, is needed to mobilize cholesterol and other lipids to expand the ER membranes so that all the virus parts will fit inside20. “When you take TMEM41B out, it has a major impact on infection,” says Vineet Menachery, a coronavirus researcher at the University of Texas Medical Branch in Galveston, who was involved in the research. The coronavirus transmembrane protein Nsp3 could also be a target: it creates a crown-like pore in the walls of the DMVs to shuttle out newly made viral RNA21.
Most viruses that have an outer wrapping, known as an envelope, form this feature by assembly directly at the edge of the cell, co-opting some of the cell’s own plasma membrane on their way out. But newly made coronavirus proteins take a different path.
For years, evidence has suggested that coronaviruses are transported out of the cell through the Golgi complex, an organelle that works like a post office, packaging molecules in membranes and sending them off to other parts of the cell. There, the virus forms a lipid envelope from the Golgi complex’s membrane; newly formed virions are then carried inside Golgi vesicles to the cell surface, where they are spat out of the cell, says virologist and cell biologist Carolyn Machamer at Johns Hopkins University in Baltimore, Maryland, who has studied coronaviruses for 30 years.
But in December, cell biologist Nihal Altan-Bonnet at the US National Heart, Lung, and Blood Institute in Bethesda, Maryland, and her colleagues reported that they had detected coronaviruses leaving the cell through lysosomes — cellular rubbish bins full of enzymes that break down cell parts22. Blocking the Golgi-based secretory pathway didn’t seem to affect the amount of infectious virus being released, says Altan-Bonnet. Her team’s evidence22suggests that viral proteins form an envelope by budding into the ER, then take over lysosomes to get out of the cell. The researchers are currently testing inhibitors that block the lysosomal exit process as potential antiviral candidates.
Leaving a cell through either the Golgi or lysosomes is slow and inefficient compared with budding out of a plasma membrane, so scientists don’t know why SARS-CoV-2 does it. Machamer suspects that the lipid composition of a Golgi- or lysosome-derived envelope is somehow more beneficial to the virus than one from the plasma membrane. “If we understood this part a little bit better, there would be great opportunities for novel antiviral therapeutics,” she says.
Last slice
On the way out of the cell, one more event makes this virus into an infectious juggernaut: a quick snip at a site of five amino acids prepares the virus to strike its next target.
Where other coronaviruses have a single arginine amino acid at the junction of the S1 and S2 subunits of the spike, SARS-CoV-2 has a line of five amino acids: proline, arginine, arginine, alanine and arginine. “Because the site was unusual, we focused on it, and it turned out that, yes, the site is essential for invasion of lung cells,” says Pöhlmann. In May 2020, he and his colleagues reported that a host-cell protein called furin recognizes and clips that string of amino acids — and the cut is “essential” for the virus to enter human lung cells efficiently23.
It’s not the first time that researchers have identified a furin cleavage site on a virus; highly pathogenic avian influenza viruses also have it, says Barclay. When a colleague sent Barclay a strain of SARS-CoV-2 in culture that had spontaneously lost the furin cleavage site, her team found that ferrets infected with this strain shed viral particles in lower amounts than did those infected with the pandemic strain, and did not transmit the infection to nearby animals9. At the same time as Barclay’s team reported its results in a September 2020 preprint, a study in the Netherlands also found that coronavirus with an intact furin cleavage site enters human airway cells faster than do those without it24.
Furin is suspected to cut the site at some point during virion assembly, or just before release. The timing might explain why the virus exits through the Golgi or lysosomes, says Tom Gallagher, a virologist at Loyola University Chicago in Illinois. “The virus, once assembled, moves into an organelle where it can be bathed in the presence of the furin protease.”
By snipping the bond between the S1 and S2 subunits, the furin cut loosens up virion spike proteins so that during cell entry they respond to a second cut by TMPRSS2, which exposes the hydrophobic area that rapidly buries itself in a host-cell membrane, says Gallagher. If spikes are not pre-clipped by furin —and they aren’t always — they bypass TMPRSS2, and enter through the slower endosomal pathway, if at all.
Two coronavirus variants, Alpha and Delta, have altered furin cleavage sites. In the Alpha variant, the initial proline amino acid is changed to a histidine (P681H) ; in the Delta variant, it is changed to an arginine (P681R). Both changes make the sequence less acidic, and the more basic the string of amino acids, the more effectively furin recognizes and cuts it, says Barclay. “We would hypothesize that this is the virus getting even better at transmitting.”
More furin cuts mean more spike proteins primed to enter human cells. In SARS-CoV, less than 10% of spike proteins are primed, says Menachery, whose lab group has been quantifying the primed spike proteins but is yet to publish this work. In SARS-CoV-2, that percentage rises to 50%. In the Alpha variant, it’s more than 50%. In the highly transmissible Delta variant, the group has found, greater than 75% of spikes are primed to infect a human cell.
Known unknowns
The scientific community is still scratching the surface of its understanding of SARS-CoV-2. Key unknowns include the number of ACE2 receptors needed to bind to each spike protein; when exactly the S2 site is cleaved by TMPRSS2; and the number of spikes needed for virus–cell membrane fusion, says McLellan — and that’s just for entry. In April 2020, a team at the University of California, San Francisco, identified at least 332 interactions between SARS-CoV-2 and human proteins25.
It is not easy to keep pace with the quickly mutating virus. Most mutations so far are associated with how effectively the virus spreads, not with how much the virus damages the host, experts agree. This month, a study reported thatthe Delta variant grew more rapidly and at higher levelsinside people’s lungs and throats than did earlier versions of the virus26.
But it is not yet certain how Delta’s mutations have supercharged the variant in this way, says Stern-Ginossar. “This is something many labs are trying to figure out.”
Our jamaican botanical wellness provides clear information on each product’s sourcing, composition, and intended benefits, empowering consumers to make informed choices about their wellness journey. With a commitment to ethical production and the highest standards, our company has become a trusted name in wellness products worldwide, catering to consumers who prioritize purity, quality, and authenticity.
As a Partner and Co-Founder of Predictiv and PredictivAsia, Jon specializes in management performance and organizational effectiveness for both domestic and international clients. He is an editor and author whose works include Invisible Advantage: How Intangilbles are Driving Business Performance. Learn more...





















1 comments:
Our jamaican botanical wellness provides clear information on each product’s sourcing, composition, and intended benefits, empowering consumers to make informed choices about their wellness journey. With a commitment to ethical production and the highest standards, our company has become a trusted name in wellness products worldwide, catering to consumers who prioritize purity, quality, and authenticity.
Post a Comment